Напророчили
Когда подтвердятся предсказания нобелевских лауреатов по химии
Если долго делать точные предсказания, когда-нибудь предсказание нагонит и вас. Трое нобелиатов по химии 2024, Дэвид Бейкер, Джон Джампер и Демис Хассабис — успешные предсказатели. Дэвид Бейкер создал Rosetta, алгоритм, который по заданной трехмерной структуре белка мог спрогнозировать необходимую аминокислотную последовательность. Джон Джампер и Демис Хассабис, наоборот, разработали нейросеть AlphaFold, способную с высокой точностью гадать по аминокислотной гуще, в какую трехмерную структуру свернется белок. Все трое, по предсказанию компании Сlarivate, стояли в этом году первыми в очереди за Нобелем. Круг пророчеств замкнулся. Правда, некоторые из них еще предстоит проверить.
У нас проблема
Дэвид Бейкер включился в гонку предсказаний первым из троицы, когда в 1993 году сменил программу философии и социальных наук в Гарварде на изучение клеточной биологии и занялся проблемой фолдинга белка. Ему хотелось понять, как аминокислотная цепочка белка сворачивается в сложную (и главное — нужную) трехмерную структуру.
Раскручиваться эта головоломка начала задолго до Бейкера: первыми Нобелевскую премию по химии за открытие трехмерной структуры белка получили еще в 1962 году Макс Перутц и Джон Кендрю. Миоглобин, дыхательный фермент мышц, стал первым белком, чью трехмерную структуру удалось разглядеть с помощью рентгеноструктурного анализа. 2500 атомов углерода, азота, кислорода, водорода и серы, соединенных в хитрую цепочку аминокислот, скручиваются в спирали, а затем эти спирали — сплетаются в корзинку вокруг железосодержащего пигмента.
Особенность этой молекулы, по словам самого Кендрю, была в ее сложности и отсутствии симметрии. Трехмерный клубок миоглобина на первый взгляд чурается закономерностей, словно бы он спутался по воле случая. Однако эта спутанность на деле оказывается и закономерной, и детерминированной. Более того, то, как выглядит трехмерный клубок, определяет его функцию: белки одной формы становятся основой мышц, рогов и перьев, другой — ферментами, третьей — порами и каналами в мембране клеток, четвертой — катушкой для наматывания ДНК.
Все это многообразие держится на скромном наборе из 20 аминокислот — звеньев, которые могут соединяться друг с другом в цепочку в бесконечном количестве сочетаний. Аминокислотная цепочка — первичная структура белка. Затем отдельные части цепочки начинают сворачиваться во вторичную структуру, образуя альфа-спирали или бета-складчатые слои. Наконец, Важно отметить, что иерархическая структура белка не соответствует последовательности стадий в процессе фолдинга. Изначально считалось, что белок действительно сворачивается последовательно: сначала в цепочке аминокислот образуются альфа-спирали и бета-слои, а уже затем из них формируется полноценная объемная структура. На самом деле образование элементов вторичной структуры и «склеивание» доменов в окончательную структуру часто происходят параллельно.
Рентгеноструктурная кристаллография, которую использовали Перутц и Кендрю, до сих пор остается золотым стандартом оценки трехмерной структуры белков. Но он не обделен недостатками: изучать структуру белков таким способом долго, дорого, и это не всегда заканчивается успехом. Например, любой белок капризен при кристаллизации и требует ручного подбора условий (некоторые — превращаться в кристалл и вовсе отказываются). Трудоемкость объясняет диспропорцию между известными аминокислотными последовательностями (база данных UniProt насчитывает 245 миллионов) и известными структурами белка (в Protein Data Bank их Небольшая поправка: на 2024 год в PDB уже около 200 тысяч структур.
Но вскоре после открытия структуры миоглобина затеплилась надежда, что кристаллография — не единственный способ узнать, в какую форму скрутится белок. Американский биохимик Кристиан Анфинсен провел серию опытов с рибонуклеазой, которая после денатурации (утраты третичной структуры) потом сворачивалась обратно в привычный клубок и продолжала кромсать РНК как ни в чем не бывало. Это означало, что структура по крайней мере некоторых белков полностью определяется аминокислотной последовательностью — и больше для правильного фолдинга ничего не требуется. Догма Анфинсена (за которую он тоже, кстати, получил Нобелевскую премию — в 1972 году) подразумевала, что знания последовательности белка в теории достаточно, чтобы узнать его трехмерную структуру. Вопрос: как? В этом и состоит проблема фолдинга.
Фактически эта уже полувековая задача сводится к трем вопросам:
- Как аминокислотная последовательность диктует трехмерную структуру белка?
- Почему и как белки могут сворачиваться так быстро?
- Можем ли мы разработать алгоритм для предсказания структуры белков на основе их аминокислотных последовательностей?
Несмотря на огромное число исследований, подтверждающих, что связь между первичной и третичной структурой белка есть, и даже предлагающих принципы их поиска, Нобелевской премии именно за эти работы так и не дали.
Здесь стоит уточнить, что второй вопрос из этой тройки: почему белок сворачивается так быстро — принципиально решен. Теория нуклеации объясняет, почему белок сворачивается в третичную структуру за миллисекунды. Сейчас с хорошей точностью можно предсказывать, например, время фолдинга и в грубом приближении пути сворачивания — с помощью простых полуэмпирических и теоретических моделей.
Но вот описание процесса фолдинга для конкретных белков с помощью моделирования, например, методом молекулярной динамики до сих пор остается слишком затратным.
Парадоксальная догма
Основная проблема задачи фолдинга — в ее катастрофической сложности, на которую почти одновременно с формулированием догмы Анфинсена указал Впервые Сайрус Левинталь сформулировал свой парадокс в докладе «How to Fold Graciously», который он представил на конференции по Мессбауэровской спектроскопии в биологических системах 1969 года. Сейчас текст доклада в интернете найти трудно, но доступна его архивная PDF-версия.
Решение этого парадокса фактически отвечает на первый вопрос проблемы фолдинга. Белок не беспорядочно перебирает все возможные варианты укладки, преодолевая сопротивление менее выгодных промежуточных вариантов укладки. Фактически задача белка сводится к задаче оптимизации, то есть поиску глобального энергетического минимума.
Этот процесс можно представить в виде движения по поверхности воронки в энергетическом ландшафте, постепенно приближаясь, по законам термодинамики, к ее дну. Положение этой ямы, как и ландшафт вокруг нее, определяется физическими свойствами аминокислот и связями, которые они способны сформировать. В процессе фолдинга аминокислотным остаткам белка доступны все возможные связи любой природы: дисульфидные мостики, водородные и вандерваальсовы связи, гидрофобные и электростатические взаимодействия и так далее. Перебирая варианты, белок в итоге оказывается на дне энергетической ямы, с которого потом практически невозможно подняться — поэтому третичная структура такая устойчивая.
Поправка
Изначально в тексте мы писали, что белковая цепь, перемещаясь по воронке, двигается в процессе фолдинга от одной промежуточной устойчивой конфигурации к другой. На самом деле, здесь модель воронки может вводить в заблуждение, поскольку таких устойчивых состояний немного, всего несколько даже для больших однодоменных белков. В 90-е были обнаружены белки, которые быстро сворачиваются в нативную форму без устойчивых промежуточных состояний — по такой сокращенной одностадийной программе работают многие небольшие однодоменные белки.
В фолдинге в этом случае белку необходимо преодолеть единственный барьер свободной энергии — переходное состояние между денатурированным состоянием и третичной структурой, которого как раз не видно в воронках. Это переходное состояние нестабильно и недолговечно, но именно оно — определяющий этап фолдинга. В этот момент у белка появляется точка нуклеации — ядро, вокруг которого затем быстро схлопывается третичная структура. То, насколько быстро белок в результате случайных флуктуаций доберется до этого энергетического барьера и преодолеет его, и определяет, какое время будет проходить фолдинг.
Как и в любой оптимизационной задаче, найти лучший путь к энергетическому минимуму довольно сложно. Поверхность этой воронки испещрена локальными повышениями и понижениями свободной энергии: холмами и впадинами, которые белку приходится преодолевать или обходить стороной. Например, можно натолкнуться на такой вариант свернутого клубка, который на первый взгляд кажется устойчивым, а на самом деле оказывается лишь лучшим вариантом из худших — локальным минимумом для в целом неустойчивой структуры.
Найти оптимальный путь до нижней точки, то есть именно ту последовательность перебора конфигураций, которую вероятнее всего проверяет сам белок, с текущим развитием вычислительных технологий не представляется возможным: например, для маленького белка в 39 аминокислот моделирование миллисекундного фолдинга занимает несколько суток работы кластеров графических процессоров. Поэтому ответ на второй вопрос: как белки могут сворачиваться так быстро — на время остается без ответа. И пока искать лучшие тропинки через эти энергетические заросли толком не удается, остается довольствоваться финальной точкой маршрута — третичной структурой белка.
Конкурс структур
Перед тем, как взяться за третичную структуру, ученые последовательно разбирались с предыдущим уровнем организации белковой структуры. То есть они пытались научиться по первичной структуре белка предсказывать вторичную: определять, где у полимера с конкретной последовательностью аминокислот будет альфа-спираль, где — бета-слой, а где — соединяющая их петля. Для решения этой задачи в 1970-х появился, например, Статистический метод Чоу — Фасмана был не первым и не единственным подходом, который появился в начале 1970-х, который позволял связать первичную структуру белка с вторичной. Похожие методы предлагали как отдельные группы: как Алексей Финкельштейн и Олег Птицын или Валерий Лим в СССР — так и международные коллаборации.
Этот метод основывался на небольшом количестве доступных на тот момент данных о последовательностях белков и их трехмерных структурах и позволял вычислить вероятности принадлежности определенной аминокислоты к определенному классу вторичной структуры. Так, аланин, глутамат, лейцин и метионин оказались «спиралеобразующими» аминокислотами, а вот на пролине и глицине, спираль, наоборот, зачастую заканчивается. Точность оригинального метода составляла тогда примерно 50–60 процентов. Затем метод чуть усложнили, добавив в вероятностное уравнение вклад аминокислот-соседей — ведь даже слабый вклад в формирование вторичной структуры каждой из соседних аминокислот мог заметно сказаться на всей структуре в целом. Это позволило поднять точность до 65 процентов.
Но худо-бедно предсказывать вторичную структуру недостаточно: альфа-спирали и бета-слои — не функциональные элементы, а небольшие строительные блоки, хоть и уже не отдельные кирпичики. Их надо еще и уложить относительно друг друга оптимальным образом. Именно для того, чтобы преодолеть этот следующий шаг, в 1994 году исследователи запустили проект Critical Assessment of protein Structure Prediction, «Критическая оценка прогнозирования структуры белка»
Раз в два года ученые стали получать доступ к последовательностям белков, структура которых уже определена рентгеноструктурной кристалографией, но держится в секрете. Задача: за несколько дней с помощью алгоритма предсказать структуру белка, пользуясь лишь его аминокислотной последовательностью, а затем — сравнить с данными кристаллографии. Чем больше процент совпадения прогнозируемой и реальной структуры — тем выше баллы. Задания различаются по сложности.
Для заданий попроще используют последовательности белков, структура гомологов которых уже известна. Если у белка есть родственник с известной третичной структурой, то ее можно использовать вместо шаблона, потому что гомологичные белки, как правило, обладают схожей структурой. В этом случае аминокислотную последовательность можно «натянуть» на известную структуру гомолога, а затем — немного подправить форму, чтобы минимизировать свободную энергию. Этот способ назвали предсказанием по шаблону (template-based). Значительно более сложная категория — предсказание структуры ab initio, сворачивание белка «с нуля», когда у исследователя на входе есть только аминокислотная последовательность и никаких известных гомологичных структур для помощи.
В 1998 году в этом конкурсе предсказателей принял участие, правда не выиграл, Дэвид Бейкер. Его оптимизационный алгоритм Rosetta на основе метода Монте-Карло относительно неплохо справился с заданиями ab initio. Rosetta брала короткие фрагменты белка с неизвестной структурой и сравнивала со схожими фрагментами в банке трехмерных структур, собирала из этих фрагментарных структур что-то наподобие чудовища Франкенштейна и оптимизировала, постепенно ведя к минимуму свободной энергии. На выходе Rosetta выдавала множество возможных решений и ранжировала их по энергетической эффективности. А поскольку такой подход требует больших вычислительных ресурсов, вскоре Rosetta вынужденно перешла с отдельных компьютерных кластеров на распределенные вычисления: авторы модели запустили проект Rosetta@home, к которому могли подключиться все желающие, чей компьютер подходит по минимальным системным требованиям.
Метод Бейкера конкретно для этой задачи подходил очень хорошо, но все-таки не идеально, — тем не менее, он каждый год оказывался среди лидеров конкурса. Среди конкурентов метода Бейкера были модели, в которых, помимо оптимизации свободной энергии, использовали еще и исследование коэволюционных последовательностей и другие подходы биоинформатики. Так было, по крайней мере, до 2016 года.
Предсказательная машина
До 2016 года включительно победители CASP не могли пробить потолок в 40 баллов из 100 возможных. Но в середине 2010-х бум нейросетей добрался и до молекулярной биологии, и в 2018 году в конкурсе предсказателей появились Джон Джампер и Демис Хассабис из компании Google DeepMind с нейросетью AlphaFold.
Через два года, усовершенствовав нейросеть до следующей версии AlphaFold2, они одержали еще более сокрушительную победу. Обе версии AlphaFold были обучены на базе данных белковых структур PDB, но помимо последовательности с размеченными физико-химическими свойствами аминокислот нейросеть использует дополнительную информацию, например, заранее узнает, что какие-то участки последовательности, вероятно, взаимодействуют друг с другом. Так, в качестве дополнительной информации служат коэволюционирующие позиции, выловленные с помощью множественного выравнивания последовательностей.
Почему такой резкий скачок произошел именно в 2018 году, N + 1 объяснил Артур Залевский из Калифорнийского университета в Сан-Франциско:
«Прорыв в качестве предсказания уже напрашивался и произошел, видимо, в первую очередь благодаря развитию железа и программных инструментов. Судя по экспериментам проекта OpenFold, количество данных для них бутылочным горлышком не было. Даже 10 тысяч структур отдельных цепей достаточно для достижения уровня качества, сравнимого с AlphaFold2, обученным на полном PDB. А в PDB достаточно данных было уже в 1999 году. Стоит, правда, оговориться, что эти условные 10 тысяч, доступные в 1999 году, были не так разнообразны, как случайно выбранные 10 тысяч белковых последовательностей из современной базы PDB. А разнообразие дает больше информации.
Ни в коем случае не хочу преуменьшать заслугу лауреатов, но они оказались в нужное время, в нужном месте и смогли максимально использовать эти возможности. Набрали команду лучших талантов: инженеров, дата-сайентистов и экспертов в структурной биологии, — выбрали четкую задачу и решили ее, не отвлекаясь на типичные академические активности».
После победы в CASP DeepMind выпустила пресс-релиз, в котором заявила, что «решила грандиозную задачу 50-летней давности в области биологии». Это, конечно, лукавство: из трех вопросов фолдинга AlphaFold отвечает лишь на последний (то есть предлагает способ предсказания структуры белков по аминокислотной последовательности), и то — в несколько упрощенном варианте.
Даже если свести этот вопрос только к построению третичной структуры и отсечь остальные стадии сворачивания белковой молекулы, задача все еще остается сложной: в ней слишком много взаимодействующих атомов. Играть в этот бисер пока бессмысленно, поэтому приходится идти на ухищрения и вместо атомов брать элементы покрупнее. В результате довольно важной для компьютерного моделирования молекулярных структур процедуры «огрубления зерна» (coarse graining) в симуляции появляются псевдоатомы — имитации молекулярных участков, для которых задают некоторые усредненные параметры взаимодействий. Участники конкурса CASP решают задачи с радикально упрощенными молекулами белка: каждая аминокислота представлялась скелетной единицей Cα — атомом углерода, из которого «растет» радикал аминокислоты. AlphaFold точно предсказывает взаимное расположение Cα атомов.
Еще одна поправка
Изначально в тексте мы писали, что полноценных достоверных подтверждений тому, что грубой структуры остова от AlphaFold достаточно, чтобы потом достроить по нему полную структуру белка, пока совсем мало. На самом деле так говорить не совсем верно: этих свидетельств, пусть чаще косвенных, к сегодняшнему дню накопилось довольно много.
Дело в том, что успешность алгоритма в конкурсе CASP оценивается именно по тому, насколько точно он воспроизвел в своем прогнозе положения остовных Cα-атомов. Поэтому первые осторожные замечания, которые изначально раздались в сторону AlphaFold, касались этого «огрубления» молекулы: будет ли его достаточно, чтобы потом достроить полную структуру белка? Но AlphaFold моделирует положение вообще всех атомов молекулы белка — и если точность моделирования остова белковой молекулы проверяли по условиям конкурса, то правильность предсказаний для положения атомов радикалов затем подтверждалась другими методами.
В частности, на точность предсказаний AlphaFold указывает успешное применение его моделей в экспериментальной структурной биологии. Например, AlphaFold дополняет рентгеноструктурную кристаллографию и помогает восстановить часть информации о кристаллической фазе, утраченной во время дифракции рентгеновских лучей, с помощью молекулярной замены. Этот метод похож на предсказания структуры по шаблону: можно взять известные данные похожих белков и на их основе «достроить» результаты рентгеноструктурного анализа нового, неизвестного белка. Вместо экспериментальных данных о белковых молекулах используют и предсказания AlphaFold. То, что структуры AlphaFold успешно справляются с этой задачей и довольно метко попадают в реальную структуру белка, подтверждает точность предсказаний и для атомов радикалов.
Несмотря на все еще неполные данные по достоверности полученных трехмерных структур белков, авторы нейросети уже собрали их базу данных, которая сейчас насчитывает более 200 миллионов структур. Это примерно в 1000 раз больше, чем доступно в PDB — базе кристаллографических структур.
Правда, пользоваться этими данными ученым приходится на свой риск: когда нейросеть предсказывает структуры неизвестных белков, никто не знает, как выглядит правильный ответ. И нельзя сказать наверняка: машина выдала правдивую структуру или галлюцинацию. Когда нейросеть играет в го или шахматы, странности игры простительны. Вопиющие причуды в игре, вроде невозможных шахматных ходов, которые делают начинающие модели, видны даже посторонним. Менее очевидные ошибки должен опровергнуть соперник (если сможет). Но в науке ошибки нейросетей, прогнозирующих, скажем, невозможные структуры белков, непростительны — в соперниках у алгоритма объективная истина.
У AlphaFold2 и AlphaFold3, которые действительно могут ошибаться, есть система маркировки участков, в которых нейросеть не уверена. Например, в этой трехмерной модели сфингомиелиназы есть участки, отмеченные синим, — в них AlphaFold уверена. А в желтых и оранжевых — сомневается:
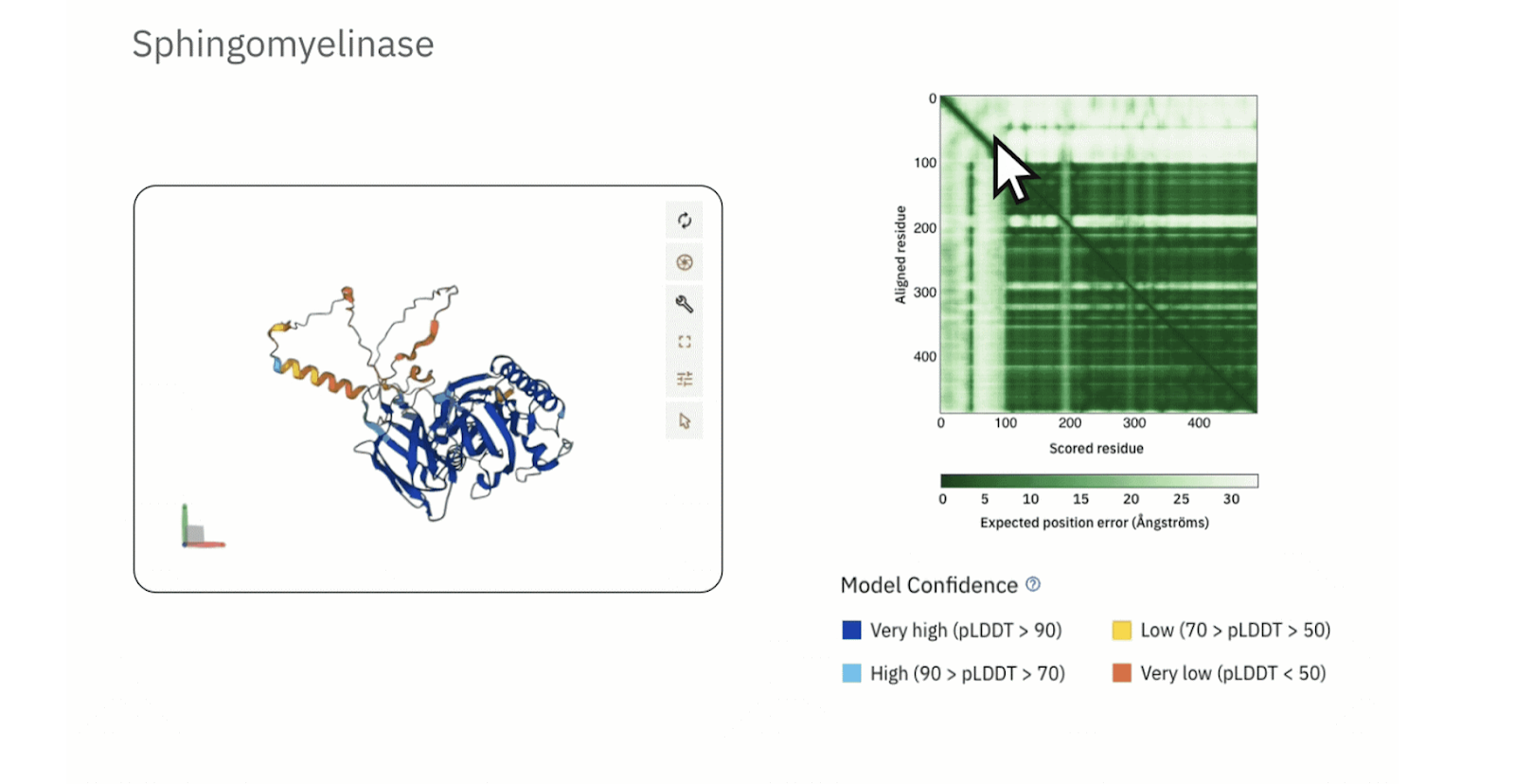
Источник изображения: European Bioinformatics Institute
С появлением в науке искусственного интеллекта и ее явным выходом за пределы академии границы и критерии того, чему можно доверять, постепенно размываются. И как освоиться в этой новой реальности — не до конца понимает и Нобелевский комитет.
Но процесс оценки эффективности идет быстро — инструмент пользуется спросом. По данным Google DeepMind, на сегодняшний день более 500 тысяч исследователей из 190 стран воспользовались базой данных AlphaFold DB.
После конкурса предсказателей исходный код AlphaFold2 был выложен в открытый доступ, что позволило сторонним исследователям тщательно протестировать его и подтвердить эффективность. Например, схожая с AlphaFold2 архитектура глубокого обучения была быстро внедрена Бейкером и его коллегами в рамках программы RoseTTA Fold.
Шиворот-навыворот
Модель Дэвида Бейкера, которая и до и после появления в игре нейросетей, хоть и держалась в самом топе конкурса, но всегда оставалась в решении проблемы фолдинга только одной из. А вот исключительной она стала в другой смежной задаче, что и отметил Нобелевский комитет.
Помимо того, что в Rosetta внедряли нейросети для увеличения точности предсказаний, модель все прошедшие годы непрерывно совершенствовали (как создатели, так и внешние пользователи) и для другого функционала, в том числе для дизайна белков — то есть поиска таких искусственных полипептидных структур, которые будут обладать заданной функцией.
Rosetta изначально разрабатывалась как универсальная программа, и калитка алгоритма работает в обе стороны: можно по аминокислотной последовательности предположить трехмерную структуру, а можно, напротив, исходя из желаемой трехмерной структуры, узнать, какая у нее должна быть аминокислотная последовательность. Благодаря этому в 2003 году лаборатория Бейкера фактически придумала белок Top7: это цепочка из 93 аминокислот, компактно уложенная в клубок из двух альфа-спиралей и бета-складчатого слоя. Совершенно новая молекула — и с точки зрения структуры, и по аминокислотной последовательности. У Top7 был один недостаток: абсолютно новый белок ничего не умел делать.
Через пять лет команде Бейкера удалось с помощью своей модели придумать и синтезировать уже более сложные белки — искусственные ферменты, у которых нет природных аналогов. Эти ферменты, которые катализировали ретроальдольную реакцию и реакцию Дильса — Альдера, пока работают хуже естественных, но их работу можно улучшить за несколько этапов направленной эволюции (за исследования этих механизмов тоже присудили Нобелевскую премию по химии, в 2018 году — подробнее о них читайте в материале «Игра в Бога»). Возможности модели продолжали расти каждый год: в 2013 году с помощью Rosetta разработали белковые структуры, которые селективно связываются со стероидами, а в 2016 — продемонстрировали возможность сборки вирусоподобной частицы.
Не поторопились?
На сегодняшний день по количеству достигнутых экспериментальных результатов и их описания в научных статьях, Rosetta выглядит основательнее AlphaFold: более 30 статей на странице проекта Rosetta против двух упоминаний высокоцитируемых статей с описанием работы AlphaFold2 и только что вышедшей AlphaFold3 в блоге проекта. DeepMind cо всеми поколениями AlphaFold обещает много: справиться с загрязнением пластика или победить голод и болезни — а в подкрепление амбициозным целям рассказывает истории исследователей, которые использовали их нейросети в своих экспериментах.
Проиграв на поле предсказания белковых структур, Rosetta тем не менее на годы опередила направление целиком именно по ценности результатов. То стремление к идеальным оптимизированным структурам белков, которое сильно замедляло и усложняло работу, пока они были неизвестны, очень ускорило процесс, когда потребовалось подгонять ответ под уже известное трехмерное решение. Главное, чтобы это идеальное решение было в голове у ученого или его можно составить из информации, которая есть в проверенной базе данных. Тогда ошибиться трудно. А если ответ заранее неизвестен, и сверить его нельзя ни с одной базой данных, то на пути к нему придется пройти через огромное число нерегулярных, неправильных и неидеальных структур.
Нобелевский комитет по химии, как и их коллеги-физики, среагировал на нейросетевой бум и оптимистично поверил в то, что создание новых вакцин, развитие экологичной химической промышленности и решение проблем устойчивости бактерий к антибиотикам благодаря достижениям нобелиатов этого года стало близко, как никогда.
Вероятно, не до конца проверенные структуры белка, созданные AlphaFold, действительно верны — и ученые на самом деле научились предсказывать структуру белка. Решение обратной задачи — предсказывать нужную для конкретной структуры аминокислотную последовательность с помощью Rosetta — тоже, похоже, часто возможно. Но как эти структуры трансформируются в функцию? Насколько уверенно белок с определенной структурой превращается в белок с определенной функцией? Пока что биология придерживается мнения, что структура определяет функцию.
Подвох, пожалуй, сейчас как раз в том, насколько хорошо мы понимаем связь между конкретной функцией и расположением конкретных атомов в пространстве. Даже к дизайну с помощью искусственного интеллекта сериновых протеаз, одного из самых изученных семейств ферментов, лаборатория Дэвида Бейкера, одного из лауреатов, подобралась только-только.
Получается, от создания Tоp7 до функционального искусственного фермента без дополнительных надстроек лаборатория Бейкера потратила 20 лет. Сколько ждать надежного и достоверного применения AlphaFold — неизвестно. Может потребоваться 20 лет, а может — 2 года. Нобелевский комитет решил этого непредсказуемого момента не ждать.
От редактора
Уже после публикации в текст было внесено несколько поясняющих уточнений и поправок.
Нейросети, кажется, научились точно предсказывать структуры белков. Структурные биологи больше не нужны?
Алгоритм машинного обучения смог предсказать структуру белка по цепочке аминокислот с точностью, которая в некоторых случаях практически полностью совпала с экспериментальными данными. Один из основателей конкурса, в рамках которого соревновались алгоритмы, уже заявил о том, что после такого проблему определения структуры белков можно считать «в определенном смысле решенной». Откуда взялся такой оптимизм, и насколько он обоснован?